婴儿腿部骨骼畸形主要表现有哪些,是MPS2吗
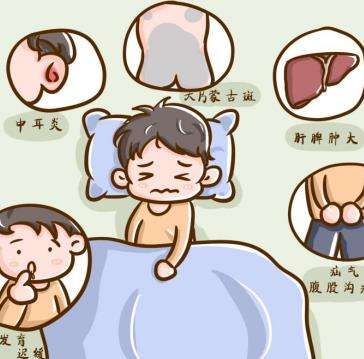
呱呱落地的新生命会引来家长亲友的诸多关注,腿部发育也是关注点之一,有的家长反应孩子总是垫着脚走路,这正常吗?婴儿腿部骨骼畸形主要表现有哪些,是MPS2吗?黏多糖贮积症2型(MPS2)是由于患儿体内溶酶体内能够降解糖胺聚糖的艾度糖醛酸-2-酯酶缺失或缺乏,而无法降解的糖胺聚糖积聚在体内引起,身体的多个器官系统都有可能受累。而且黏多糖贮积症2型是一种X连锁隐性遗传疾病,是由于母亲X染色体上的IDS基因突变导致的,如果母亲是携带者,那么所生的女儿有50%的概率成为携带者。患者外观和骨骼的异常是相似的,患者通常在出生时表现正常,最常见的表现特征是面部特征变粗,在2-4岁之间变得明显。关节挛缩是早期出现的症状,尤其指骨关节挛缩。患者由于关节挛缩导致行动受到限制。由于关节僵硬,脚后跟紧,MPS2患者通常用脚趾走路。MPS2患者往往有宽厚的鼻子,鼻子张开。嘴唇肥厚,舌头增大凸出。整个人生阶段,头围都很大。患者的身高在婴幼儿时期往往比正常同龄孩子更高,但是到了4-5岁,患者的身高开始落后于同龄孩子。患儿常见舌大,腺样体和扁桃体肥大,咽喉中GAG沉积会导致声音嘶哑。患儿牙齿形状不规则,常见牙龈组织增生及牙源性囊肿。患儿可出现传导性和感觉神经性听力损伤,听力下降并伴有耳部反复感染。在这里建议家长朋友留心观察,如果发现宝宝有相关的异常表现建议找当地三甲儿童医院儿科内分泌遗传代谢科就诊,酶学检测加尿GAG检测即可明确诊断是否是黏多糖贮积症II型。黏多糖贮积症II型的标准治疗是酶替代疗法,患儿确诊后,宜尽早开始治疗。通过阅读上文,想必大家对于“婴儿腿部骨骼畸形主要表现有哪些,是MPS2吗”也有了答案,婴儿时期与家长的相处时间比较多,也是最容易发现宝宝的问题的时候,尤其
2021-11-18