小孩上呼吸道阻塞的常见原因是什么,是亨特综合征吗
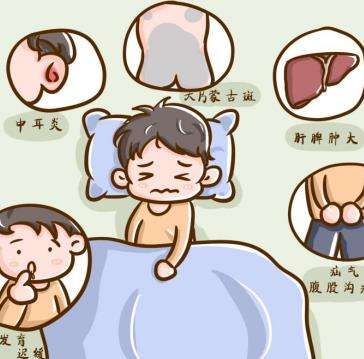
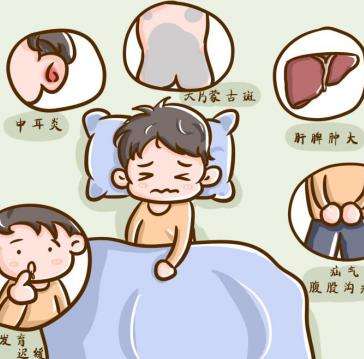
上呼吸道阻塞常见呼吸困难,呼吸费力,小孩上呼吸道阻塞的常见原因是什么,是亨特综合征吗?小孩上呼吸道阻塞的常见原因是什么,是亨特综合征吗?小儿由于舌体相对较大,咽部发育尚不完善,较成年人来说狭窄,呼吸肌发育不完善,肺容量小,所以容易导致呼吸道阻塞的病症,还有误吸入异物也容易呼吸道阻塞的发生,排除这些因素,那么就要考虑是疾病因素导致的了。罕见病亨特综合征(黏多糖贮积症2型,MPS2)的患儿会有上呼吸道阻塞的表现,除此之外,还有反复耳部感染、频繁呼吸道感染、打鼾、呼吸急促、呼吸困难、不断流鼻涕、经常咳嗽感冒、钉状牙、牙齿排列稀疏、听力障碍等耳鼻喉方面的异常表现。黏多糖贮积症2型(MPS2)是一种罕见的渐进性致残、致死的X连锁隐性遗传病。是由于艾度糖醛酸-2-酯酶缺失或缺乏,导致糖胺聚糖无法降解,患者体内贮积的糖胺聚糖可累及多个系统,关节活动受限是黏多糖贮积症2型的重要表现之一,还有可能出现典型面容,表现为头大、额头突出、宽鼻、厚唇等。除了外观特征的改变,患儿出生时生长表现正常,生长速率随着年龄的增长而降低,几乎所有的儿童在青春期前都表现出生长迟缓,身材矮小。如果遇到家里的宝宝出现上述的情况,建议找当地三甲儿童医院儿科内分泌遗传代谢科就诊,酶学检测加尿gag检测即可明确诊断。酶替代疗法是治疗黏多糖贮积症2型的标准疗法,患者确诊后,宜尽早开始酶替代治疗,早期酶替代治疗可减缓疾病进展,改善患者预后。通过上文的讲述,相信大家对于“小孩上呼吸道阻塞的常见原因是什么,是亨特综合征吗”都有所了解了,家长平时要小心呵护宝宝,发现宝宝有罕见病的相关表现要及时就诊治疗。
2021-11-18